渐冻症术语是肌萎缩侧索硬化(ALS),也称运动神经元病(MND),是一种累进性神经退行疾病,以控制肌肉收缩的上、下运动神经元选择性退化和凋亡为特征,引发瘫痪、最终导致死亡,患病率约为4-6/10万。大约10%的ALS病例是家族型的,90%是散发型的。从发现SOD1基因突变到现在,共确定了30余种致病基因,其中C9orf72,FUS,TARDBP,SOD1等比例较高,而EPHA4,ATXN2等20余种基因比率较低。肌萎缩侧索硬化症病因涉及兴奋毒性、氧化应激、神经营养因子、自身免疫、中毒、感染等,目前无有效的治疗方法,仍然是 “不治之症”。
已发现的30种致病基因和修饰基因与ALS的病理发生有关,其机制可归纳为细胞表面受体、RNA加工、蛋白质合成、能量代谢、内质网功能、轴突转运等几个方面的异常。其中C9orf72和EPHA4分别参与了RNA加工、内质网、细胞膜受体三种ALS的发病机制。 C9orf72(chromosome 9 open reading frame 72)蛋白与细胞运输和自噬有关,敲低斑马鱼的C9orf72表达可干扰神经分支的形成和缩短运动神经元轴突,推测C9orf72表达水平降低影响了神经元内细胞运输。
ALS动物模型是研究ALS的病因、病理、发病机制和治疗的重要工具。目前建立ALS的啮齿类动物模型主要是SOD1、TDP-43、VCP、FUS等基因的转基因或基因敲除小鼠,Wobbler自发突变小鼠,不同模型病理表现不同。目前常用的啮齿类动物模型只反映了10%左右的病因,需要研制覆盖更广泛病因的基因修饰动物模型。
大鼠在生理、行为、神经等研究最常用的实验动物,大鼠神经系统疾病模型不仅有好的行为学表现,也能表现一些在小鼠模型上不能表现的病理改变。我们推测C9ofr72基因修饰大鼠模型将比小鼠有更好的表型。中国医学科学院医学实验动物研究所基因动物工程平台张连峰团队建立了C9ORF72基因敲除大鼠(图1)。相关研究工作2021年3月发表在FEBS J杂志。

初步研究发现,C9orf72敲除单独不足以诱发ALS表型。利用低剂量兴奋性毒性诱导剂红藻氨酸(KA)皮下注射后,敲除大鼠出现进行性运动功能障碍(图2)与运动神经元的丢失(图3);高尔基复合体断裂和异常的囊泡运输(图4),提示C9orf72敲除大鼠需要额外的兴奋性毒性的刺激来诱发ALS。C9orf72功能的丧失和兴奋性毒性协同作用是诱发ALS发病的重要因素。而在临床上,肌萎缩性脊髓侧索硬化症(ALS)的大多数病例中都发现了运动神经元兴奋毒性的改变。作为一种可能的肌萎缩性脊髓侧索硬化症的常见介质,它可以与其他遗传因子相互作用,触发疾病的发病机制。
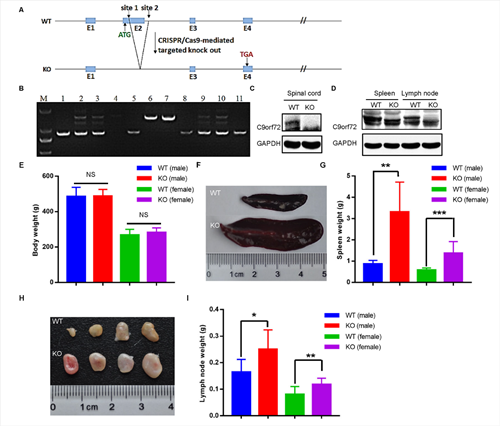
图1. C9ORF72基因敲除大鼠的建立
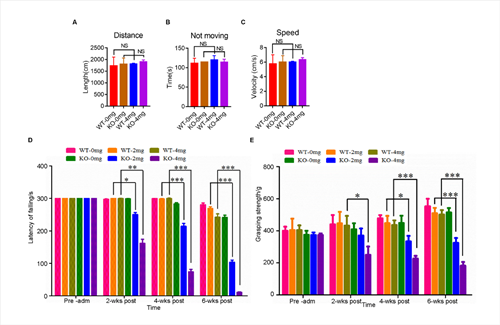
图2. C9ORF72基因敲除和兴奋毒性共同诱导运动缺陷
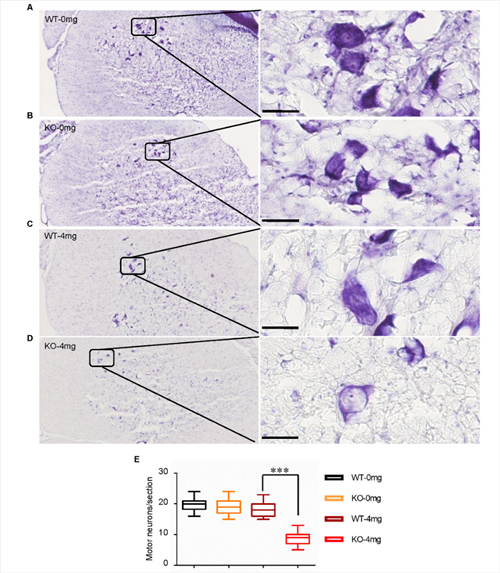
图3. C9ORF72基因敲除和兴奋毒性联合作用导致运动神经元丢失
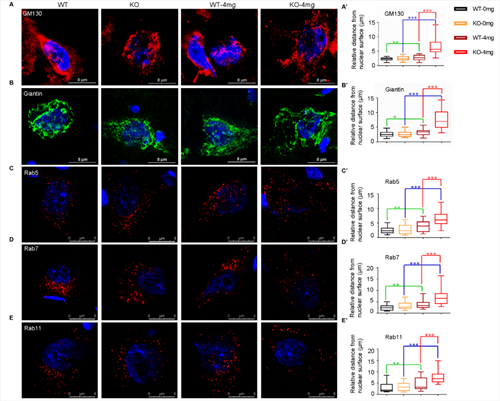
图4. C9ORF72基因敲除和兴奋毒性联合作用导致运动神经元中高尔基复合体的脆弱性增加
有4篇C9orf72 BACs转基因小鼠模型的报道,都产生RNA foci和DPRs(dipeptide repeats),其中两个模型未出现ALS/FTD(frontotemporal dementia,额颞痴呆)的神经退行性变或行为特征;一个模型表现出FTD的特征,另一个表现出与ALS和FTD相似的运动障碍和神经退行性变。这些数据提出了一个问题,即产生RNAfoci和DPRs的C9orf72六核苷酸重复序列扩增是否足以导致疾病的发病。在人类患者中,G4C2六核苷酸重复序列扩增显著抑制C9orf72蛋白表达,C9orf72基因敲除导致斑马鱼运动障碍,提示C9orf72功能丧失可能是ALS发病的一种特殊机制。
该项大鼠模型的建立及初步研究结果表明,C9orf72单独敲除不足以引起ALS,但增加了运动神经元对其他危险因素如兴奋毒性的敏感性,并在体内以协同方式引起ALS;也提示C9orf72功能丧失可能是ALS发病机制之一,提高运动神经元中C9orf72水平可能是一种治疗策略。
参考文献
 京公网安备 11010502043083号
京公网安备 11010502043083号